
In spring 2010, there will be two documents manifesting the requirements with regard to clinical data for the certification of medical devices: the Directive 2007/47/EC as well as the MEDDEV 2.7.1 Rev. 3.
This Med-Info may be used as an introduction to the structure and some essential details regarding the requirements for clinical data, but it does not obviate the need for review of the original documents.
Directive 2007/47/EC
With regard to the requirements for clinical data, 2007/47/EC now includes a variety of details –in contrast to the original 93/42/EEC Directive. Although the relevant contents are not really new, this Directive:
• provides a definition of clinical data, which is understood as “safety and/or performance information that is generated from the use of a device”; in order to generate adequate data, these may be taken from clinical investigations, scientific literature, or other clinical experience with equivalent devices
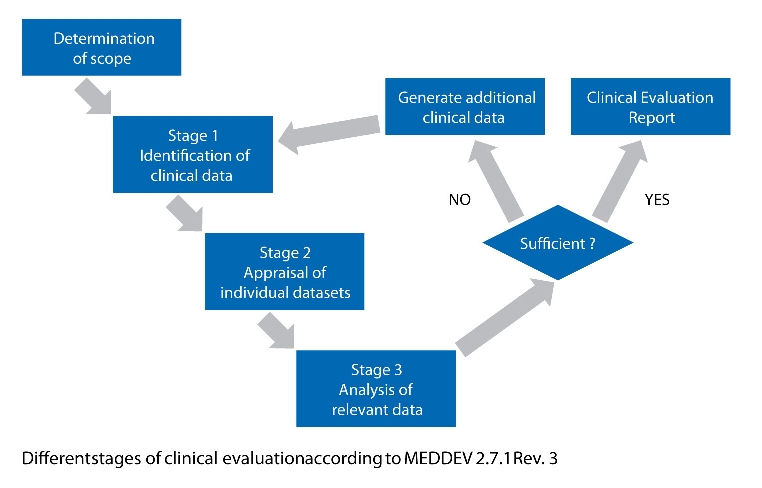
• makes a clinical evaluation for any medical device mandatory; such an evaluation must follow “a defined and methodologically sound procedure”. Furthermore, the clinical evaluation shall be documented. An additional explicit requirement in that context is an active update of the performed clinical evaluation with data from post-market experience
• postulates a positive risk-to-benefit ratio based on clinical data
• sets out different clear conditions in case the literature route is followed:
a) The evaluation of the relevant scientific literature must include a total of four different aspects, i.e. safety, performance, design characteristics as well as intended purpose of the device, which means that this route will only be feasible if it covers adequate information for all of these different aspects
b) Equivalence of all data taken for demonstration of the essential requirements must be demonstrated
c) In addition, the evaluation must be performed “critically”
• requires due substantiation to base the demonstration of essential requirements for any device on “performance evaluation, bench testing and pre-clinical evaluation alone”
• includes different explicit requirements in case the clinical trial route is followed:
a) The results of all investigations made are to be evaluated “critically”
b) Clinical investigations are to be performed “unless it is duly justified to rely on existing clinical data” for implantable devices and class III devices
c) Different specific requirements are dealing with the procedure of a clinical trial, including reporting of serious adverse events and end of a study, communication requirements in case a clinical trial is refused/halted by a Member State, or modification is requested; besides, the documents to be provided for a clinical investigation are set out
• requires due justification where post-market clinical follow-up as part of the post-market surveillance plan is not deemed necessary
• postulates an adequate justification in case clinical data are not deemed necessary, based on risk management output, under consideration of the specifics of the device/body interaction, the clinical performances intended, and the claims set out. In that, such a process should also be documented adequately; eventually this again implies a clinical evaluation
• provides a modification of classification of medical devices, i.e. a new definition of central circulatory system
For details, reference is made to Article 1, Article 15, Annex VIII, Annex X and Annex XI of the Directive.
MEDDEV 2.7.1 Rev. 3
The new MEDDEV 2.7.1 Rev. 3 regarding clinical evaluation should be regarded to be more than just an update of the previous revision, as it now additionally includes almost the full GHTF SG5/N1R8 “Clinical Evaluation” guideline as well as an extensive clinical evaluation checklist for Notified Bodies.
The following sections will not list the entire content of the MEDDEV paper, but will rather focus on aspects which are either new, or anticipated to result in issues for different processes. Additionally, basic information regarding general requirements may be taken from our earlier Med-Info “Requirements for Clinical Data” (August 2007).
The preface of MEDDEV 2.7.1 Rev. 3 points out that it is not legally binding; nevertheless “it is anticipated that the guidelines will be followed within the Member States”. An alternative approach has to be duly justified in either case.
Therefore, a sound rationale should be part of the documentation.
<